|
Stratospheric Processes And their Role in Climate
|
||||||||
| Home | Initiatives | Organisation | Publications | Meetings | Acronyms and Abbreviations | Useful Links |
![]()
|
Stratospheric Processes And their Role in Climate
|
||||||||
| Home | Initiatives | Organisation | Publications | Meetings | Acronyms and Abbreviations | Useful Links |
![]()
A.R. Ravishankara, NOAA, Boulder, USA (ravi@al.noaa.gov)
Organizing Committee:
R. A. Cox, Cambridge, UK, IGAC representative
M. J. Kurylo, NASA/NIST, Washington, DC, USA, NASA and SPARC representative
A. R. Ravishankara, NOAA, Boulder, CO, USA, SPARC representative
K. A. Wolfe, CSC, Boulder, CO, USA
There have been a large number of field measurements that have unearthed interesting compositional information that suggests the need for additional data on gas-phase reactions, photochemical processes, and heterogeneous/multiphase reactions that occur within the UT/LS. From a chemist’s point of view, this region (Figure 1) is a challenge because of the low temperatures accompanied by significant pressures, the presence of condensed matter, and highly variable levels of ozone and water vapour. The laboratory research community in kinetics and photochemistry met in October 1996 in Strasbourg (France) to take stock of current progress in our understanding of heterogeneous and multiphase reactions (see SPARC Newsletter N°9, 1997, pp. 6-15). However, the state of gas-phase kinetics and photochemistry had not been assessed for a long time. Therefore, a workshop was held during 22-27 July 2001 in Breckenridge (Colorado) to assess the state of gas-phase and photolytic parameters relevant to the above issues.
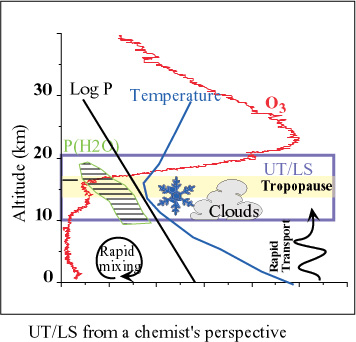 |
| Figure 1. Chemist's view of UT/LS |
|
(For a better resolution of the images, please click on the plot or contact the SPARC Office)
|
SPARC, IGAC, NASA, NOAA, EC, NERC UT/LS, and EC-IPO sponsored this workshop, which brought together a few key atmospheric modellers and field measurement scientists with researchers from the laboratory community. The specific areas that were covered were:
The topic of short-lived compounds and their impact on UT/LS was included because it is a major topic of the 2002 WMO/UNEP Scientific Assessment of Ozone Depletion and it was thought important to air out the chemistry issues involved in this topic as input for the assessment activities.
The workshop programme consisted of scene-setting talks, followed by extensive discussions. There were two opportunities to present posters, which were displayed throughout the workshop period. All attendees were encouraged to bring a few view graphs of their results to aid a comprehensive discussion, and most attendees took advantage of this opportunity.
The following summaries highlight the discussions and findings of this workshop in the seven areas listed above and are based on the rapporteurs' reports. These reports were done in collaboration with the speakers and the session chairs.
The scene setting talks in this session were given by R. Salawitch (NASA/JPL) and P. Wine (Georgia Institute of Technology). The session chair was P. Wennberg (California Institute of Technology) and the rapporteur was P. Monks (University of Leicester).
Photodissociation of atmospheric molecules by solar radiation plays a fundamental role in atmospheric chemistry. The photodissociation of trace species such as ozone and formaldehyde contributes to their removal from the atmosphere, but probably the most important role played by these photolytic processes is the generation of highly reactive atoms and radicals from stable molecules. Photodissociation of trace species and the subsequent reaction of the photoproducts with other molecules are the prime initiator and driver for the bulk of tropospheric chemistry.
The first order rate coefficient for the photolytic destruction of a molecule is given by the expression,
 |
|
(1) |
where s is the absorption cross-section (cm2), f
is the quantum yield for the destruction of the molecule, F is the wavelength dependent solar flux density (photons cm-2 s-1),
and T is the temperature. If we are interested in a specific process, the quantum yield will be for that specific outcome following the absorption
of light by the molecule under consideration. For example, in the photolysis of NO2
| NO2 + hn -> NO + O(3P) |
|
(2) |
the rate of removal of NO2 is given by
|
|
(3) |
where the photolysis frequency, j2, is the first order rate coefficient for the removal of NO2 and F is the quantum yield for the dissociation of NO2.
The solution of equation (1) under both the physical and chemical conditions encountered throughout the UT/LS lies at the heart of understanding photolytic processes and their role in the chemistry of these regions. The photolysis rate or j value can be either measured in the atmosphere or computed using parameters measurable in the laboratory (s, F) and the actinic flux, which is computed from either extraterrestrial flux or observations. In either case, high quality laboratory measurements are required to evaluate the factors in equation (1) to design and validate measurements of j values.
The current state of our information on the actinic flux was discussed and some recommendation for future work was made. They included the following research activities:
(a) The evaluation of the O3 cross section as a function of T,
(b) The resolution of the differences in the absolute values of s(O2) in the Herzberg continuum,
(c) Recommendation of a top-of-the-atmosphere flux, which takes into account both seasonal and solar cycle effects for use in modelling studies,
(d) Atmospheric transmission measurements in the O2 Schumann-Runge absorption region to test model parameterisations of this spectral region,
(e) Atmospheric measurements of O (3P) as a critical check on ozone photolysis rates.
Following the discussions on the actinic flux, the available laboratory data were reviewed and the following issues of high priority for UT/LS were noted:
(a) Extension of the absorption cross section measurements to longer wavelengths,
(b) Extension of the absorption cross section measurements to typical UT/LS temperatures,
(c) Extension of quantum yields to wavelength regimes (and temperature/ pressure as appropriate) where atmospheric photodissociation actually occurs in the UT/LS,
(d) Assessment of the low energy (electronic) transitions,
(e) Assessment of the missing transitions,
(f) Assessment of the metastable states,
(g) Assessment of the weak absorbers,
(h) Assessment of the overtone transitions,
(i) Assessment of the H2O complexes.
These suggestions were followed by discussions of the experimental factors involved in such research as well as issues such as parameterisation of the cross sections, extrapolation and interpolation of laboratory data, and the limitations and needs of such undertakings. It was further noted that a number of criteria must be considered when evaluating the atmospheric importance of a given photolysis frequency. These include composition with respect to lifetime (production vs. loss routes), the role of the photoproducts, the influence of clouds and aerosols, the sensitivity of the action spectrum to changing values of s and f, and the variation of the action spectrum and atmospheric sensitivity with respect to changing solar zenith angle.
The scene setting talks in this session were given by J. Troe (University of Gottingen), D.M. Golden (SRI International and Stanford University), and H. Hippler (University of Karlsruhe). The session chair was J. Troe (University of Gottingen) and the rapporteur was J. Barker (University of Michigan).
This was a very lively session with a great deal of debate. The discussions clearly showed the emerging approaches to addressing this important , but complex, area of kinetics.
Chemical species from the planetary boundary layer, the middle troposphere, and the middle stratosphere are found near the tropopause and thereby influence the composition of the UT/LS. Conditions there are such that a wide variety of pressure-dependent reactions may be important. This is both because of the large number of organic species important in the troposphere and because of the low pressures and temperatures that occur near the tropopause. In this session, pressure-dependent reactions were categorised into several classes. Some experimental and theoretical tools used for determining the rate constants needed for accurate atmospheric models were subsequently described. The protocols currently in use for analysing and tabulating conventional fall-off data for recombination and decomposition reactions were discussed. Finally, a summary of future needs for determining, evaluating, and tabulating pressure-dependent reactions important in the UT/LS were listed.
Reactions are pressure-dependent when the rate of collisional energy transfer is competitive with the rate of a chemical process that depends on internal energy. Even "inert" gases like argon (Ar) and nitrogen (N2) participate in energy transfer. Since the rate of energy transfer depends on the total pressure (including "inert" gases), the over-all reaction is pressure-dependent. The following classes of reactions were noted and discussed:
(a) Recombination/Decomposition Reactions: These reactions are important in all regions of the atmosphere. In recombination reactions, chemical energy is released and a highly vibrationally excited product is produced. If energy transfer is too slow, the excited product re-dissociates and there is no net reaction. If energy transfer is very fast, then the excited product is efficiently stabilised. The fall-off curve is a quantitative representation of the efficiency of stabilisation as a function of the total pressure. Decomposition reactions are the reverse of recombination reactions: a stable molecule is activated by collisions and the highly vibration ally excited molecule decomposes. The efficiency of the collisional activation is described by the fall-off curve. Fall-off curves for recombination and decomposition reactions for the same chemical system are connected by microscopic reversibility and thus are described by the same physics and can be represented using the same functional forms. Reactions of this type are currently included in the NASA/JPL and IUPAC data evaluations. An example of such a reaction is the recombination/decomposition reaction:
NO2 + NO3 <-> N2O5 (4, -4)
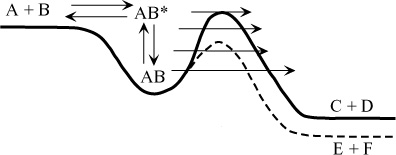
(b) Chemical Activation Reactions: In this general class, a bimolecular reaction leads to a single, highly vibrationally excited intermediate that is capable of further decomposition, isomerisation, or collisional stabilisation (Figure 2). The product branching ratio depends on pressure because collisional stabilisation of the excited intermediate results in an increased yield of the stabilized intermediate at the expense of decomposition or isomerisation products. Reactions of this type are not currently included in the data evaluations in a systematic way.
Figure 2. A reaction proceeding through the formation of an adduct (For a better resolution of the images, please click on the plot or contact the SPARC Office)
An example of such a reaction, which is included empirically in data evaluations, is the extremely important reaction of OH with CO.
OH + CO <-> HOCO* (5a, -5a) HOCO* -> H + CO2
(5b)
(c) Chemical Excitation Reactions: This is a reaction sequence that has found renewed importance under atmospheric conditions. An example of such a sequence is the reaction between the RO2 radical and NO, where a product (alkoxy radical) is left with a large amount of internal energy so that it can undergo other reactions (for example, decomposition).
RO2 + NO -> RO* + NO2 (6a) RO* -> decomposition products (6b) RO* + M -> RO + M
(6c)
(d) Multi-Well, Multi-Channel Reactions: This class includes reactions in which vibrationally excited species are produced by chemical activation, thermal activation, photoactivation, or some other process, and then undergo competitive reactions via several pathways with rates that depend on the amount of excitation. Collisional deactivation slows the energy-dependent reaction rates and hence causes changes in branching ratios, etc. Reactions of this type are not currently included in the data evaluations. Free radical isomerisation/decomposition reactions fall into this category. In the atmosphere, alkoxy radicals can undergo decomposition and isomerisation reactions, which depend on vibrational energy, as well as bimolecular reaction with O2 (it is not known whether this bimolecular reaction depends on vibrational energy). Another example involves nitrate formation in the reaction of alkyl peroxy radicals with NO.
RO2 + NO <-> ROONO* (7a, -7a) ROONO* -> RO + NO2 (7b) ROONO* -> RONO2* (7c) RONO2* + M -> RONO2 + M
(7d)
Theory has played, and will continue to play, a major role in describing, deriving, and extrapolating association/dissociation reaction rate coefficients. In this endeavour, electronic structure calculations continue to be very useful for elucidating reaction mechanisms and identifying transition state structures. As computational capabilities have improved, the accuracy of calculated thermochemical quantities has approached that needed for chemical kinetics calculations for high temperature combustion systems. In contrast, the low temperatures found in the UT/LS require thermochemical accuracy that can only be achieved for a few small molecular systems (and then, only with great difficulty). To make useful chemical kinetics predictions for the UT/LS, considerable progress will be necessary in developing computing resources and computational tools. Recent calculations on the reactions OH + HONO2, ClO + OH, and RO2 + NO illustrate the usefulness of such electronic structure calculations. Several possible transition state structures for the reaction of OH + NO2 have been identified by ab initio calculations, but some can be eliminated because their predicted isotope effects contradict experiments. Branching ratios for the ClO + OH reaction have been calculated based on calculated transition state properties.
In addition to the ab initio calculations, theoretical treatment of reaction kinetics and dynamics is an important venue for better atmospheric data. The study of the dynamics of elementary reactions leads to improved theoretical rate constants. Variational Transition State Theory and the Statistical Adiabatic Channel Model were developed as a result of fundamental investigations on theoretical reaction dynamics. These theories provide an essential connection between macroscopic elementary rate constants and the fundamental dynamics on ab initio potential energy surfaces. The theories give good insight into the expected behaviour of observed elementary rate constants and hence they are valuable as background for evaluating experimental data.
Master equations that describe the simultaneous interplay of energy-dependent collisional energy transfer and energy-dependent chemical reactions are necessary for the most accurate descriptions of pressure-dependent reactions. Master equations of varying complexity are needed for the various classes of energy-dependent reactions listed above. The most complex pressure-dependent systems involve multiple potential energy wells and multiple reaction pathways. Although several methods have been developed for solving the resulting master equations, each method has strengths and weaknesses. Considerable work is needed to increase the speed and accuracy of the existing methods and to identify which methods are most suitable for certain classes of problems.
Semi-empirical formulations (for example of Troe, Oref, Frenklach, and others) exist for the purpose of analysing, evaluating, and summarising experimental fall-off data for single-channel, single-well unimolecular/recombination reactions. The formulation published by Troe is the most popular of these, although further evaluation is needed to determine whether it is the most accurate (Figure 3). These formulations are far more practical for data analysis than master equation calculations, and they are of good general accuracy. The use of semi-empirical formulae by the NASA/JPL and IUPAC evaluation panels is discussed below.
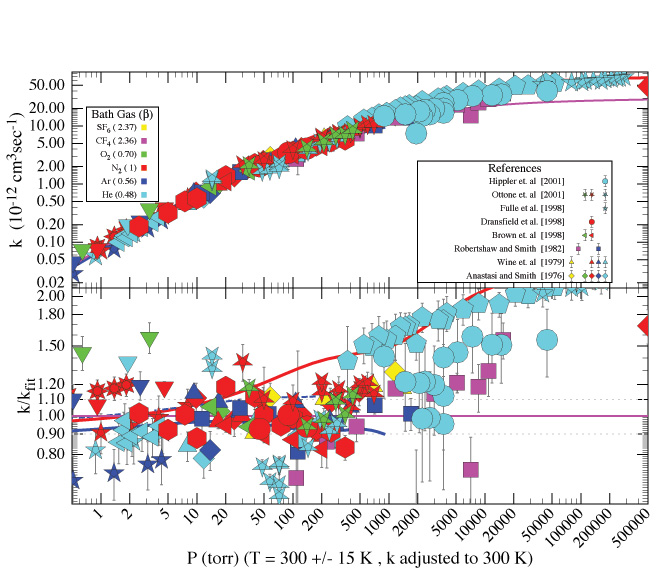 |
| Figure 3. Fall-off curve for the reaction of OH with NO2 |
|
(For a better resolution of the images, please click on the plot or contact the SPARC Office)
|
Data evaluation is an essential component in atmospheric chemistry research. Multiple investigations of the same reaction are needed to establish reproducibility and to cover the wide ranges of temperature and pressure important in the atmosphere. Inevitably, the various data sets are of uneven quality and the experimental results must be spliced together in order to obtain complete understanding of a reactive system. Data evaluation panels' function by compiling data from different sources, analysing the similarities and differences, and making informed judgments of relative quality. The end result is a set of recommendations that must be communicated to all interested parties. In the atmospheric science community, the recommended rate constants are adopted by modellers for baseline comparisons, as well as by experimentalists. In this way, the atmospheric science community attacks problems more efficiently, using the best available current data. Data evaluation is the final essential step necessary for efficient utilisation of the resources spent on laboratory measurements. Currently, the evaluation panels summarise bimolecular reactions, recombination (and decomposition) reactions, and a few complex bimolecular reactions, as well as photochemistry and heterogeneous reactions. The pressure dependence of the complex bimolecular reactions is currently approximated as a linear function, while the fall-off of the recombination reactions is treated in more detail. Troe's semi-empirical formulation is the basis for both the NASA/JPL and IUPAC evaluations of experimental fall-off data, although the details differ in the ways the two evaluation panels have chosen to apply the formulations.
At the workshop, several key questions were articulated and discussed. It was concluded that further experimental and computational work could help answer the questions:
(1) What is needed for improved data on recombination reactions?
(2) Is the high pressure limiting rate coefficient, k, independent of T?
(3) Is the rate constant for vibrational relaxation a useful surrogate for k
(4) How important is the radical-complex (chaperone) mechanism for energy transfer and radical-radical recombination? Can it be predicted? How can it be incorporated into master equation calculations?
(5) Should the NASA/JPL data evaluation protocol for representing pressure fall-off be changed?
(6) Should the NASA/JPL data evaluation panel develop protocols for other classes of pressure-dependent reactions?
The scene setting talks in this session were given by R. Atkinson (University of California, Riverside) and L. Jaegle (University of Washington, Seattle). The session chair was C. Zetzsch (Fraunhofer Institute, Hannover) and the rapporteur was G. Tyndall (National Center for Atmospheric Research).
The presence of VOCs (Volatile Organic Compounds) in the lower troposphere has long been recognised as necessary for the production of ozone. However, their role in the overall chemistry of the UT/LS is less certain. VOCs in the UT/LS could impact the formation of odd-hydrogen radicals, the production and/or enhancement of aerosols, and in situ ozone formation. The study of VOC oxidation at low temperature is not as complete as might be desired, partly due to experimental difficulties encountered when handling low-volatility compounds at reduced temperature.
A thorough discussion of the current state of rate coefficients and product data was carried out. The database for the reactions of OH, Cl, NO3, and O3 with alkanes, alkenes and oxygenated hydrocarbons were reviewed. The case of OH reaction with acetone was highlighted and it was revealed at this meeting by Orlando et al. that the reaction of OH with acetone was unlikely to produce acetic acid.
A discussion of peroxy radical reactions followed. It was noted that the recent SPARC-IGAC evaluation of peroxy radical reactions (see http://www.sparc.sunysb.edu/html/tyndall/index.html) discussed most of the issues involved in these processes. In addition to HO2 and CH3O2, other peroxy radical reactions may play a role in the UT/LS, such as the reactions of acyl peroxy radicals with other peroxy radicals. Consensus has only recently been reached on the values of the rate coefficients for some of these reactions, while the products have been the subjects of only limited study at temperatures other than 298 K. The reactions of HO2 and CH3O2 with acetyl peroxy, CH3C(O)O2, are both thought to have minor channels to form acetic acid at room temperature. The occurrence of these channels in increasing amount at low temperature could be very important with regard to aerosol composition in the UT/LS.
It was concluded that better and more widely applicable detection methods need to be developed for both peroxy and alkoxy radicals. Most rate coefficients for peroxy radicals have been measured by UV absorption spectrometry, which is not very sensitive or specific for a given radical. Alkoxy radicals have the advantage that they can be detected by laser-induced fluorescence; however, this technique becomes increasingly less sensitive as the pressure and temperature increase, and also as the size of the molecule increases. A number of laser techniques appear promising, and mass spectrometric techniques should also be investigated. In addition, methods for the detection of stable reaction products should be investigated. In this respect, interactions with the field measurement community may be invaluable. Continued collaboration between experimentalists and theoreticians should further improve our understanding of reaction mechanisms at the molecular level, which will assist in the prediction of reaction rates in regimes that are difficult to access in the laboratory.
Based on modelling efforts and field measurements, it was shown that short-lived species could be transported vertically in the atmosphere via rapid convection and play a major role in remote areas such as the upper troposphere. A related issue is the range of lifetimes of species transported to the UT/LS by convection. Some of the products of isoprene oxidation have lifetimes of several hours, and it is possible that such compounds, as well as brominated and iodinated hydrocarbons, can be transported locally to the UT by convective activity. This requires a much more detailed speciation in models than is usually included.
The roles of organic aerosol were also discussed. It was noted that to predict the composition and growth rates of aerosols at low temperatures, a suite of parameters for relevant organics are necessary. These include vapour pressures, solubilities in sulfate solution of the appropriate strength, and uptake coefficients (all at temperatures encountered in the UT/LS).
The scene setting talks in this session were given by J. Rodriguez (University of Miami) and S. Sander (NASA/JPL). The session chair was R. Friedl (NSAS/JPL) and the rapporteur was D. Rowley (University College, London).
Halogens play a key role in the UT/LS region of the atmosphere due to their participation in catalytic cycles that destroy ozone. Since halogenated source gases in the atmosphere include both natural and anthropogenic compounds, understanding the photolytic release of halogen atoms from these compounds, and the subsequent gas phase chemistry is central to elucidating the role of halogen chemistry in the ozone budget. The halogen families of particular interest in the UT/LS are chlorine, bromine and iodine. Fluorine is not considered to play a significant role in the atmosphere due to its inefficient release from the source gases and the rapid formation and high stability of the inactive reservoir species HF. The bonding strengths of the halogen atom and the reactivity of halogenated free radicals dictate, along with the magnitude of the atmospheric source, the role of other halogens in the atmosphere.
A detailed discussion of the current state of halogen reactions was conducted. It was noted that recent measurement campaigns have greatly improved the ability to test gas phase and heterogeneous chemistry schemes against atmospheric composition. For chlorine in particular, simultaneous measurements of HCl, ClONO2, ClO and Cl2O2 from aircraft and balloon observations have been reported from recent campaigns such as POLARIS, SOLVE and THESEO-2000. While POLARIS data showed that total Cly correlated well with N2O abundances, as would be expected for long-lived chlorine source gases, data from the SOLVE campaign indicated that the sum of individual inorganic chlorine species abundances was lower than the expected total inorganic chlorine content based upon decomposition of source gases. For bromine, far fewer data have been obtained on the abundances of the key species. BrO measurements indicate concentrations between 10 and 20 pptv in the LS, and thereby confirm the involvement of Br in ozone loss. HBr measurements of ca. 1.5 pptv however, are systematically not reproduced in models. This has led to speculation that the BrO + HO2 reaction and/or the OH + BrO reaction produce HBr via minor reaction channels. The presence of iodine in the UT/LS region is as yet unconfirmed by measurements. While the first attempts to measure iodine abundances (as IO) in this region essentially reported zero abundance, more recent work has indicated IO abundances between 0.65 and 0.80 pptv in the LS.
Laboratory studies of many of the key kinetic and photochemical parameters for halogens have been reported. However, a number of these studies exhibit serious disagreements, and further work is required with improved techniques and methodologies. Individual rate-limiting reactions for important atmospheric ozone loss cycles, and reactions controlling halogen partitioning were discussed individually, along with recommendations for further study.
It was noted that many 'new' techniques have been adopted from other fields of research. In addition, data from chemical dynamics studies, such as state-resolved measurements of differential cross-sections, can be used to calculate thermal rate constants and to provide mechanistic insight into reactions and their temperature and pressure dependencies. Theoretical studies are also contributing increasingly to the database on gas phase halogen chemistry. Ab initio studies allow calculation of energies of reaction intermediates and transition states. RRKM-type calculations using these energetics can then be used to calculate rate constants.
The scene setting talks in this session were given by M. Ko (AER) and P. Seakins (University of Leeds). The session chair was R.A. Cox (University of Cambridge) and the rapporteur was J. Burkholder (NOAA, Aeronomy Laboratory).
A comparison of atmospheric transport times versus chemical lifetimes suggests that any compound with a globally averaged lifetime of less than 30 days would be called a short-lived compound. Examples of short-lived compounds are alkenes, some alkanes, oxygenates, some haloalkanes, and certain sulfur (SO2, CS2) and nitrogen (ROxNOx) species. The session was limited to haloalkanes and sulfur species, which are a focus of the 2002 WMO/UNEP Ozone Assessment.
Atmospheric model studies (2D and 3D) have demonstrated the importance of including short-lived compounds and their atmospheric degradation products in the calculation of ozone depletion potentials (ODP). The scope of short-lived compounds is however not limited to the evaluation of ODPs but may impact the stratospheric sulfate layer as well as the HOx and NOx budgets.
There are three major issues associated with short-lived compounds.
(1) Short-lived source gases are not well mixed in the troposphere and therefore cannot be treated in model calculations in the same way as long-lived compounds such as CFC-11 and CFC-12.
(2) Model results are dependent on location and time of emission (variation of OH field and actinic flux with season) of the short-lived compound making it difficult to define a single-valued ODP.
(3) Stratospheric residence times and tropospheric washout lifetimes are an important parameter in model calculations but are currently poorly defined.
The implications of the short lifetimes are that the degradation pathways and the formation of intermediate products need to be evaluated for each source gas on an individual basis. A guide to the degradation pathways is obtained by comparison with the more comprehensively studied analogous alkanes. Laboratory measurements of OH rate coefficients, photochemical processes, and heterogeneous processes for source gases and degradation products under UT conditions are needed for this evaluation. The emphasis in this session was placed on the evaluation of haloalkanes that have been observed in the atmosphere (CH3I, CH2ClI, C2H5I, CH2I2, CH3CHICH3, and CH3CH2CH2I) along with 1-bromopropane, a proposed future emission. All the processes that need to be evaluated to estimate the effect of short-lived compounds on stratospheric ozone are shown in Figure 4.
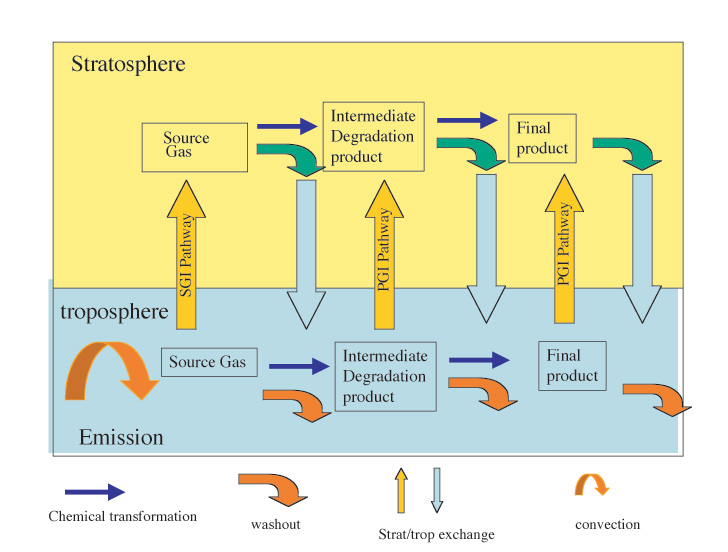 |
| Figure 4: The pathways by which halogens in a short-lived molecule can reach the stratosphere |
|
(For a better resolution of the images, please click on the plot or contact the SPARC Office)
|
In addition to the haloakanes, sulfur species are also important short-lived compounds. The formation of aerosol particles in the UT and subsequent transport to the LS can change the sulfate surface area without changing mass burden. Convection may change how particles are formed in the UT. Laboratory studies of the degradation pathways and formation of aerosol particles under UT conditions are required. These studies include, but are not limited to, the evaluation of the degradation pathways for dimethyl sulfide (DMS) and the formation of sulfuric acid aerosol under UT conditions.
The scene setting talks in this session were given by D. Fahey (NOAA, Aeronomy Laboratory) and N. Donahue (Carnegie-Mellon University, Pittsburgh). The session chair was M. Kurylo (NASA and NIST) and the rapporteur was A. Wahner (KFA-Juelich).
Nitrogen oxides play important roles in ozone production and destruction in the UT/LS regions and influence the chemical composition and reactivity of UT/LS aerosols. Kinetic data needed to improve our understanding in these roles were identified at the workshop. In the reaction of OH with HNO3, which is one key reaction that controls NO2 and HNO3 abundances and provides a sink for lower stratospheric HOx, more studies at UT/LS temperatures (< 230 K) are needed. A better theoretical understanding of the origin of temperature and pressure dependence would also be useful. In the reaction of OH with NO2 (Figure 3), which is another key reaction for NO2 and HNO3 abundances, the possible production of HOONO at low temperatures and the value of the rate coefficient for the channel that produces HNO3 are uncertain; they need to be investigated further. Also of interest is the production of peroxy nitrates, ROONO, in the reaction of RO2 with NO and the pressure and temperature dependence of this production.
Heterogeneous chemistry of the UT/LS has many unexplored reactions because the area is relatively new compared to gas-phase studies and because of the chemical complexity in the UT/LS. The studies of the N2O5 heterogeneous loss reactions provide an interesting example of the inherent complexity of the heterogeneous process. The reactive uptake is lower on crystallised aerosols than on liquid aerosols and much lower on nitrate-containing aerosols (up to a factor of 50) or on aerosols with an organic coating (5 to 10 times). Especially for the UT, this detail for the N2O5 heterogeneous reaction is important because of the role of N2O5 as a reservoir of NOx.
Observational studies in the UT/LS have provided substantial detail and coverage for the speciation and sources of the reactive nitrogen reservoir, NOy. In the LS, most of the NOy reservoir is comprised of only a few species and variability is low. In the UT, the complexity is increased by the presence of organic source gases. Budget studies in both regions have been successful in accounting for the principal NOy component species. Global coverage in NOy measurements is very sparse overall. Most measurements have been made of NO and NO2. Satellite observing systems are not available yet for UT/LS measurements of NOy. The first commercial aircraft data for NO has proven to be of value.
Accurate rate constant for the reactions involving NOy species are essential for maintaining progress in key UT/LS issues. Leading issues include calculations of the ozone tendency, the production and loss balance of NOx in the UT, and the partitioning of HOx and NOy reservoirs. The study of the NOx/NOy ratio in the summer lower stratosphere stands as an important example of how improved accuracy in fundamental rate constants improves our confidence in LS photochemistry.
The Heidelberg NOx/NOy workshop in March 2000 discussed in detail many modelling, laboratory, and observational issues related to nitrogen oxides in the UT/LS. The value of possible future reaction rate studies was prioritised.
The workshop report is available on the web at: http://www.aero.jussieu.fr/~sparc/News18/18_Fahey.html.
The scene setting talk in this session were given by M. Pilling (Leeds University). The session chair was C. Kolb (Aerodyne Research) and the rapporteur was V. Orkin (NIST).
It is clear that laboratory measurements of kinetic and photolytic processes remain key area of research in atmospheric chemistry. The future needs for atmospheric modelling purposes demand increasing accuracy and precision in photochemical data. Data evaluation, an important element of the kinetic support for atmospheric studies, places additional demands on laboratory data of good quality. Therefore, a discussion of the role of laboratory studies used to obtain such parameters was carried out during this workshop.
It was concluded that the reliability of evaluations would be improved if experimental data were always made available in the original scientific publications together with a clearly described uncertainty analysis. Furthermore, every attempt should be made to conduct laboratory measurements over pressure and temperature ranges representative of the real atmosphere. This is particularly important for termolecular reactions as well as for bimolecular reactions that exhibit non-Arrhenius behaviour at low temperatures. Also, in some cases, measurements over the relevant range of water vapour pressures are critical.
The measurements of branching ratios and reaction products are very important for correct/complete modelling and should be conducted in concert with absolute rate constant measurements. Similarly, measurements of the rate constants for the reactions of isotopically labelled compounds can help in the determination of the reaction channels and, thus, can be of atmospheric importance.
The collaboration and inter-comparison of the results obtained via laboratory studies, theoretical calculations, atmospheric modelling, and field measurements are essential for further progress in understanding the chemistry of the atmosphere. Such collaboration can also provide information on the accuracy of laboratory measurements of certain rate constants. Sensitivity analyses of modelling studies can also help prioritise reactions for further laboratory study as well as define required improvements in the accuracy of reaction rates and photolysis parameters. Finally, various experimental methods used to obtain kinetics and photolytic data were described and discussed, and future measurement methods that are coming on line were described.
![]()