Previous: Method of calculation Next: Conclusions Up: Ext. Abst.
Results and Discussion
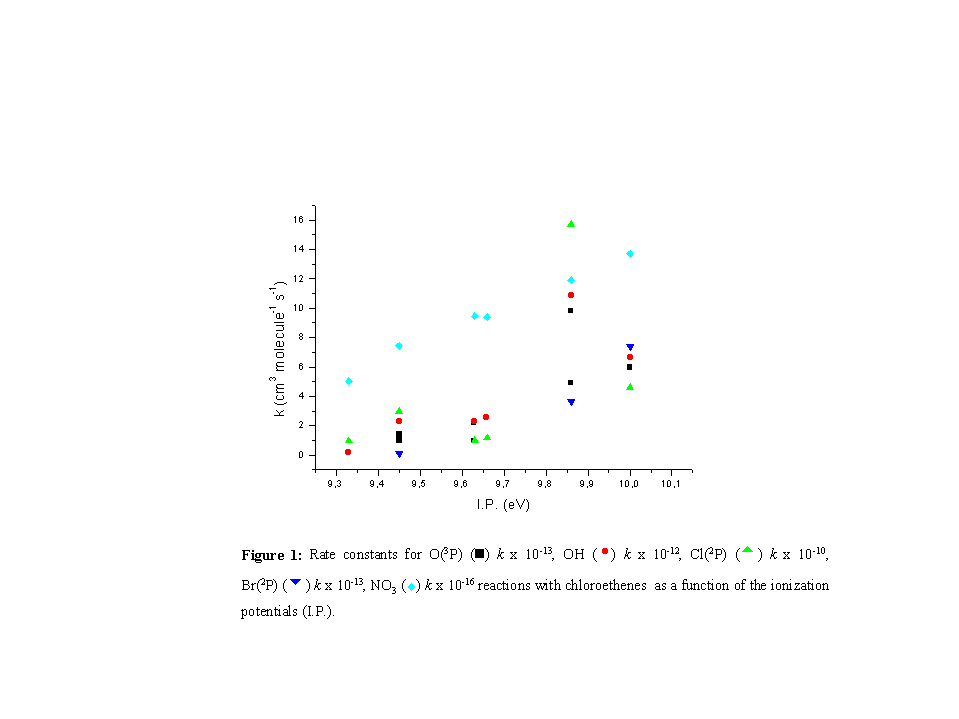
The reactivity in the alkene homologous series, as has been pointed out, correlates strongly with the ionization potential of the alkene [4]. Thus, it is generally found that the overall rate constant, k, at room temperature, for the addition reaction of the electrophile to the alkene, increases as the I.P. of the alkene decreases. However, in the present study an opposite reactivity trend was found thus the reaction rate constants increase as the I.P. of the corresponding chloroethenes increases [5] (Figure 1).
In the molecular-orbital approach, frontier-orbital interactions
generally determine the barrier heights and reaction rates in
radical or atom-molecule reactions [6]. A frontier orbital treatment
is based upon the interaction of the frontier molecular orbital
of the molecule (HOMO) with the frontier molecular orbital of
the radical or atom (SOMO or LUMO). The higher the energy of the
HOMO (low I.P) or the lower the energy of the LUMO (High electron
affinity), the greater the propensity for reaction, as represented
by the following equation:
k (cm3 molecule-1 s-1) ª 1 / (ESOMO EHOMO) ª deltaE
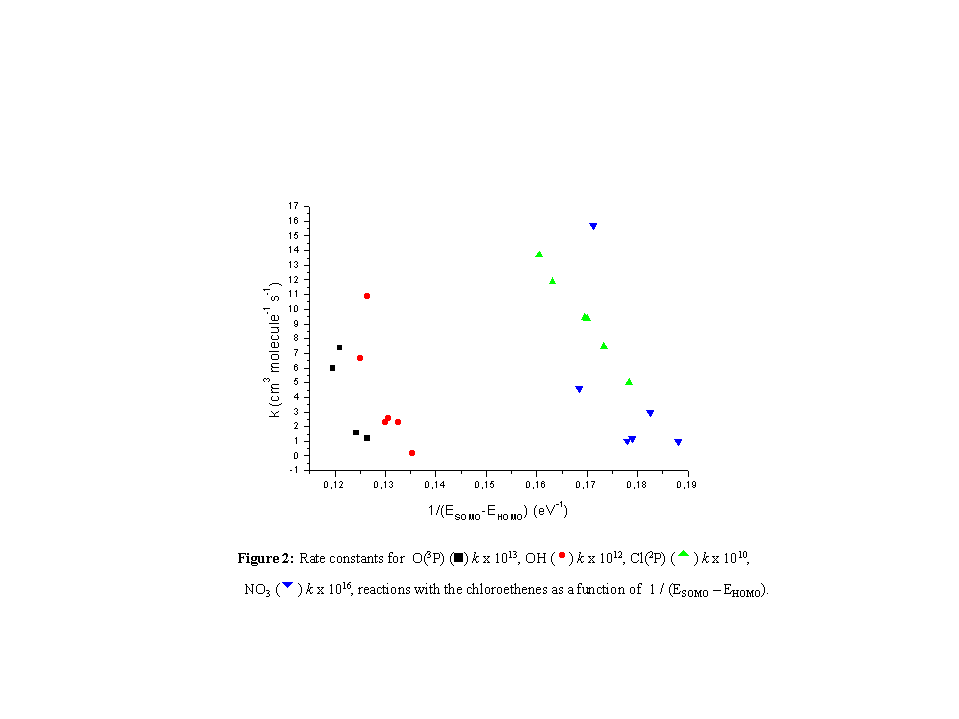
It can be observed in Figure 2 that for the reactions with the
chloroethenes in this series this correlation is not obeyed. In
fact, the reacting trend shows just the opposite, i.e., for a
higher value of deltaE, a lower rate constant is observed.
However, considering the different interaction probabilities of
the electrophiles with the chloroethenes, which are represented
according to the Frontier Orbital Theory by the HOMO coefficients
or more exactly in this kind of reactions, by the second order
perturbation (e") [6], a good correlation is found between the experimental k values and the corresponding reactivities. The following equation
relates the perturbation with the energies of the HOMO and LUMO
frontier orbitals.
e" = (cHi cSj bij )2 / (ES-EH)
where cHi is the atomic orbital coefficient of atom i in the HOMO of the
chloroalkene, cSj is the coefficient of the atomic orbital of
the electrophile in the SOMO, bij is the resonance integral between
atoms i and j.
Furthermore, the trend for the chloroethenes is similar with the
different electrophiles (Figure 3), as the rate constants increase
with the increasing perturbation.
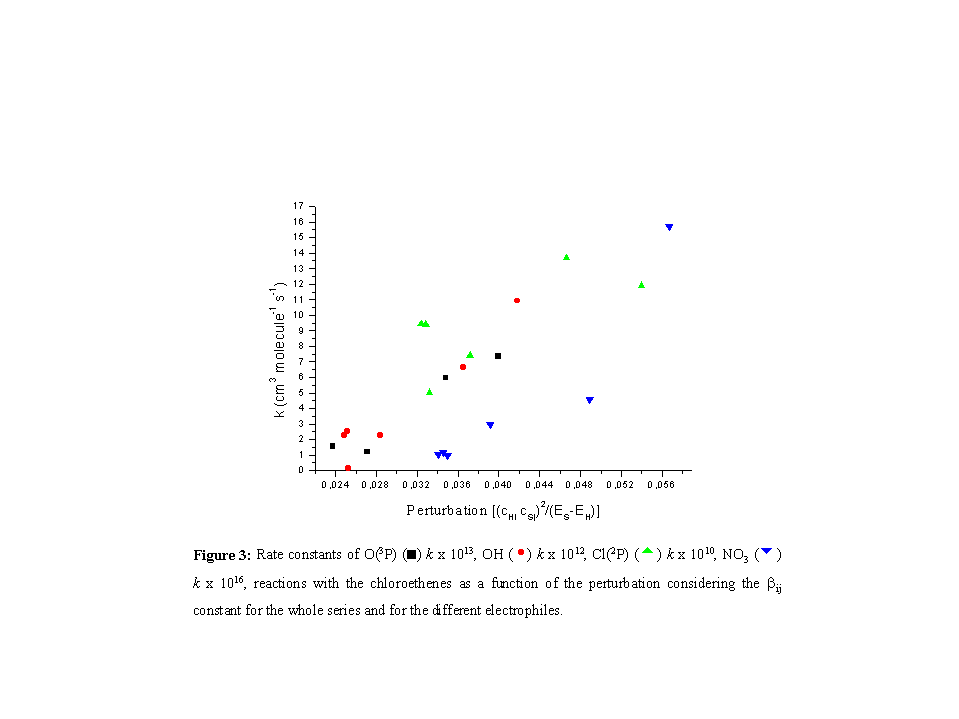
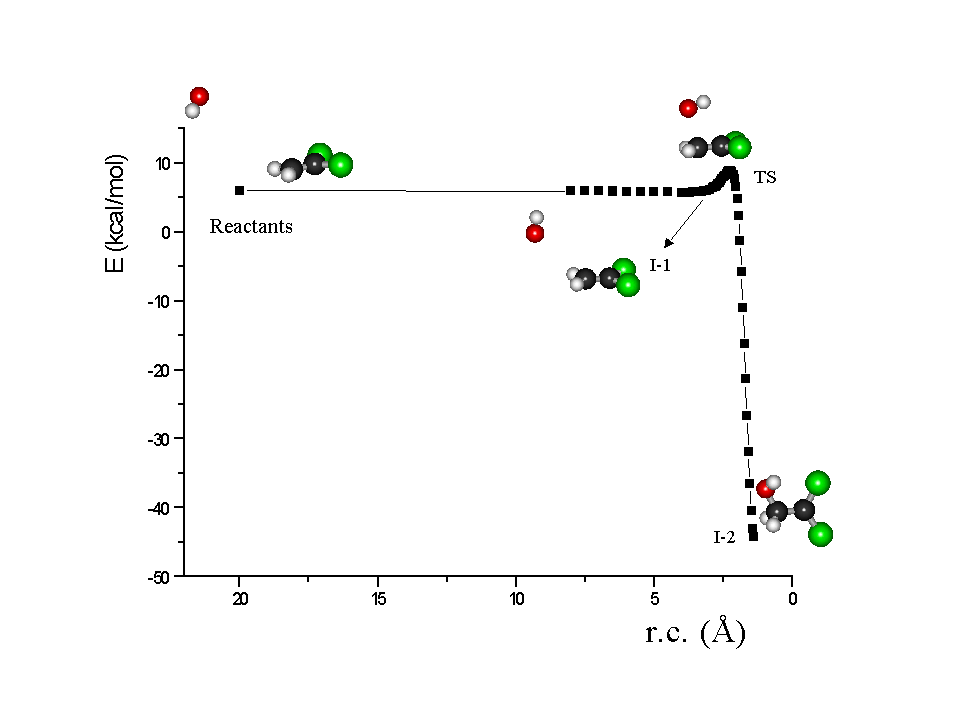
Figure 4: Reaction Coordinate for the reaction of OH + CH2=CCl2 as an example, calculated by PM3 method, where I-1 is a long-range
complex, T.S. is the corresponding Transition State and I-2 is
the stable covalent adduct or Intermediate.
From the computational analysis of the reaction pathways, we have
estimated the activation energies (Ea) and the total energies
(DHr) of the proposed reactions (Figure 4). It was not our aim,
however, to determine accurate activation energies for each reaction.
The reaction pathways were studied using semiempirical, PM3, and
ab-initio calculations (at UHF and B3LYP level of theory, with
6-31G** basis set). With these semiempirical calculations it is
possible to observe, with Br, Cl and OH but not with O and NO3,
the presence of a long-range complex (I-1) prior to it been collisionally
quenched to form a stable covalent adduct (I-2). A similar trend
for the Ea and DHr was observed between the semiempirical and
ab-initio calculations
Table 1: Activation Energies (Ea) (kcal mol-1) and Heat Contents (DHr)
(kcal mol-1) calculated by
PM3 method for the addition reaction of different electrophiles
to chloroethenes.
|
|
|
|
|
|
|
|||||||
| Ea | deltaHr | Ea | deltaHr | Ea | deltaHr | Ea | deltaHr | Ea | deltaHr | Ea | deltaHr | |
| O(3P) | 3.68 | -62.07 | 3.59 | -64.48 | 4.49 | -66.39 | 4.56 | -65.96 | 4.31 | -52.07 | 5.29 | -56.61 |
| OH | 3.76 | -47.84 | 3.07 | -50.20 | 4.74 | -49.69 | 4.71 | -49.25 | 4.21 | -50.63 | 4.31 | -50.52 |
| Cl | 0.10 | -37.15 | 0.03 | -39.05 | 0.50 | -34.05 | 0.94 | -34.44 | 0.81 | -36.74 | 1.10 | -35.12 |
| Br | 0.05 | -25.25 | 0.37 | -27.70 | 0.49 | -23.29 | 0.18 | -23.23 | 0.47 | -24.52 | 1.45 | -23.82 |
| NO3 | 3.16 | -43.98 | 3.37 | -43.19 | 2.86 | -39.29 | 2.73 | -38.60 | 0.22 | -40.83 | 1.53 | -38.41 |
In Table 1 the activation energies for the addition reactions
of the different electrophiles to the chlorinated ethenes are
shown. The Ea, in general, increase with the increasing chlorine
substitution, although for 1,1 dichloroethene, which is the most
reactive chloroethene, the addition reaction has the lowest Ea
value.
Calculated heats of reaction DHr are also presented. All the addition
reactions studied are exothermic, with DHr generally decreasing
with increasing chlorine substitution.