INFIQC Facultad de Ciencias Químicas Universidad Nacional de Córdoba Pabellón Argentina, Ciudad Universitaria 5000 Córdoba - Argentina
FIGURES
Abstract
Introduction
Chloroethenes are produced in large amounts and are widely used in industrial activities as solvents, dry cleaning, degreasing agents and in the PVC production. As highly volatile organic compounds, significant fractions enter the atmosphere. In the troposphere the reaction with OH radicals is the dominant primary oxidation step and also the reaction with the chlorine atom should be considered since the rate constant for this process is larger than for the reaction with OH. The possibility of Br-atom reaction occurring in the troposphere has been suggested [1]. The reaction with O(3P) atoms is generally not competitive with the other paths because of the low concentration of O(3P) atoms although it is a prototype system for the addition reactions to the C=C bond. The NO3 radical has strong oxidizing capacity and although it generally much less reactive than OH, its peak tropospheric concentration is higher during the night, making its reaction with the halocarbons comparable to that with OH radicals [2].
The total atmospheric burden of a halocarbon is determined both by the
rate of its release and by the atmospheric lifetime (itself directly related
to the rates of loss processes). Photolysis in the stratosphere (and in
the upper troposphere) constitutes are important sink for the halocarbons.
The atmospheric lifetime against photolysis is shorter for molecules that
contain relatively more chlorine than fluorine and is even shorter if bromine
is substituted for chlorine. Molecules that contain hydrogen in place of
halogens are susceptible to attack by OH in the troposphere, as are molecules
possessing double bonds.
The reactions of CH2=CHCl, CH2=CCl2, Z-CHCl=CHCl, E-CHCl=CHCl, CHCl=CCl2 and CCl2=CCl2 with O(3P), Cl(2P) and Br(2P) atoms and with OH and NO3 radicals were studied in this work using semiempirical and ab-initio calculations.
Method of calculation
The geometry of reactants and the corresponding HOMO energies were calculated using semiempirical methods, AM1 and PM3 from the MOPAC 7.0 program and ab initio calculations at HF and B3LYP levels of theory with the 6-31G** basis set, using the Gaussian-98 suite of programs.
The ionization potential (I.P.) of the chloroethenes was also obtained at the same levels of calculation noting that the PM3 method agrees better with the experimental values. The rate constant values considered were an average between those obtained in our laboratory and those taken the from literature [3] using different experimental methods.
Results and Discussion
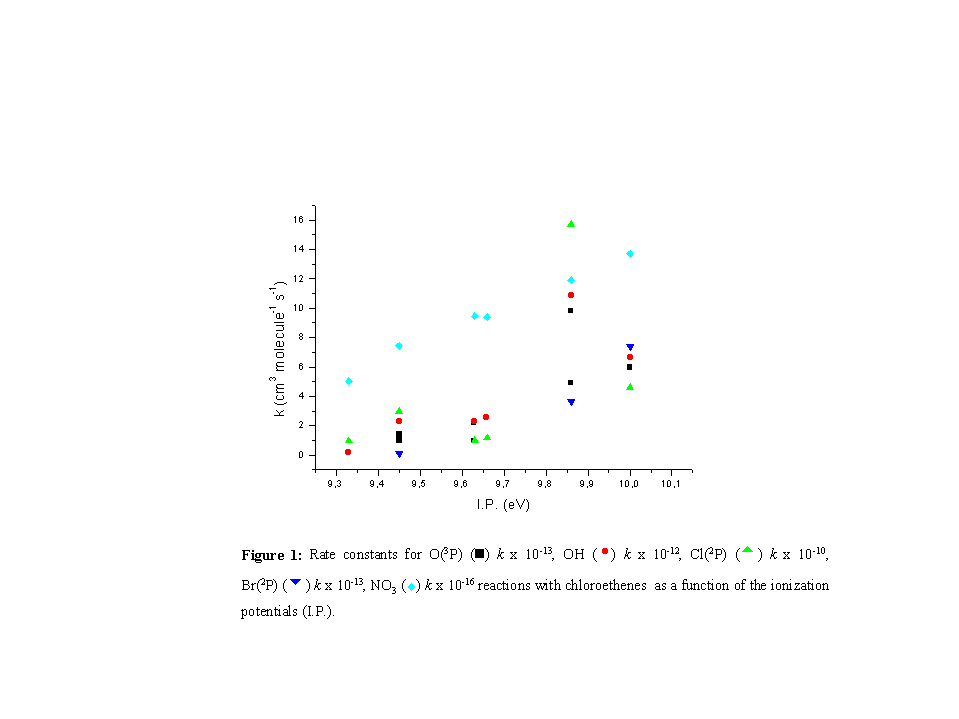
The reactivity in the alkene homologous series, as has been pointed out, correlates strongly with the ionization potential of the alkene [4]. Thus, it is generally found that the overall rate constant, k, at room temperature, for the addition reaction of the electrophile to the alkene, increases as the I.P. of the alkene decreases. However, in the present study an opposite reactivity trend was found thus the reaction rate constants increase as the I.P. of the corresponding chloroethenes increases [5] (Figure 1).
In the molecular-orbital approach, frontier-orbital interactions generally
determine the barrier heights and reaction rates in radical or atom-molecule
reactions [6]. A frontier orbital treatment is based upon the interaction
of the frontier molecular orbital of the molecule (HOMO) with the frontier
molecular orbital of the radical or atom (SOMO or LUMO). The higher the
energy of the HOMO (low I.P) or the lower the energy of the LUMO (High
electron affinity), the greater the propensity for reaction, as represented
by the following equation:
k (cm3 molecule-1 s-1) ª 1 / (ESOMO
EHOMO) ª deltaE
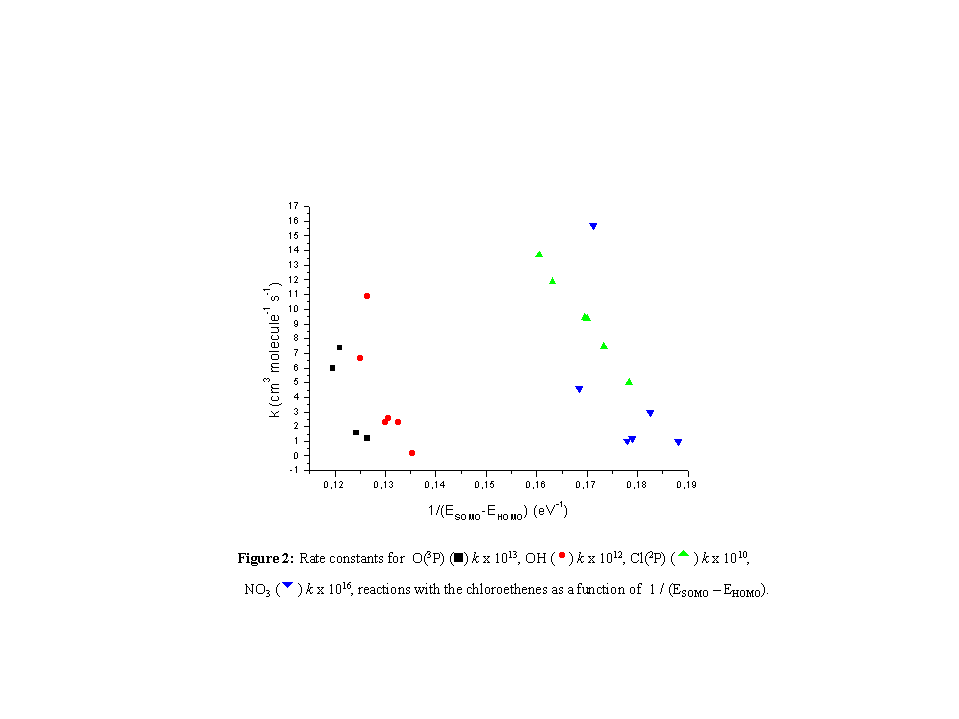
It can be observed in Figure 2 that for the reactions with the chloroethenes
in this series this correlation is not obeyed. In fact, the reacting trend
shows just the opposite, i.e., for a higher value of deltaE,
a lower rate constant is observed.
However, considering the different interaction probabilities of the electrophiles
with the chloroethenes, which are represented according to the Frontier
Orbital Theory by the HOMO coefficients or more exactly in this kind of
reactions, by the second order perturbation (e") [6], a good
correlation is found between the experimental k values and the corresponding
reactivities. The following equation relates the perturbation with the
energies of the HOMO and LUMO frontier orbitals.
e" = (cHi cSj bij
)2 / (ES-EH)
where cHi is the atomic orbital coefficient of atom i in the
HOMO of the chloroalkene, cSj is the coefficient of the atomic orbital
of the electrophile in the SOMO, bij is the resonance integral between
atoms i and j.
Furthermore, the trend for the chloroethenes is similar with the different
electrophiles (Figure 3), as the rate constants increase with the increasing
perturbation.
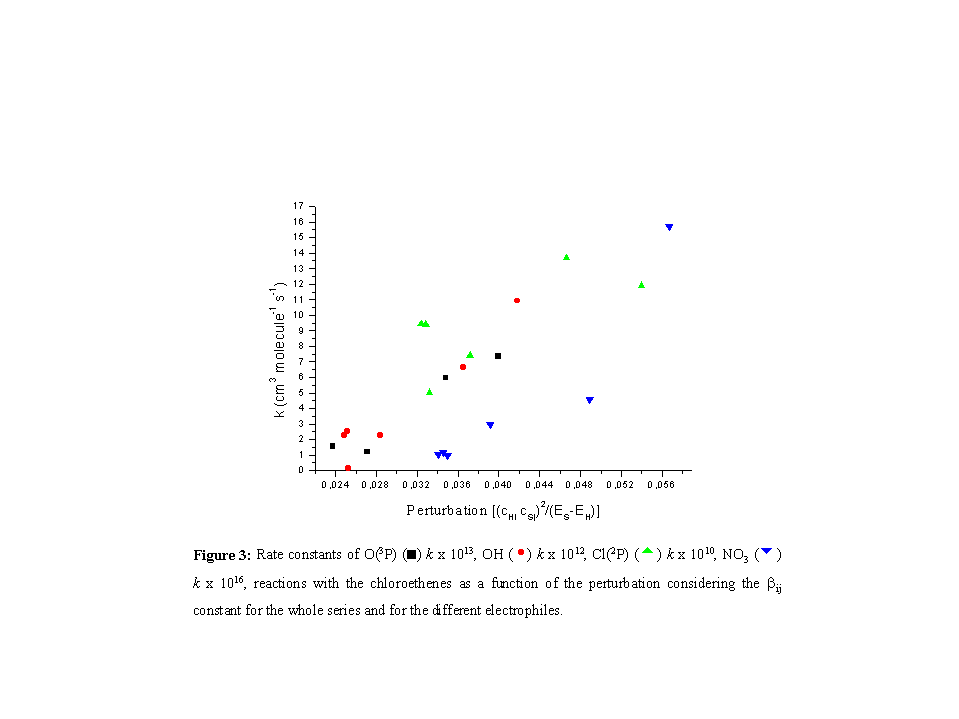
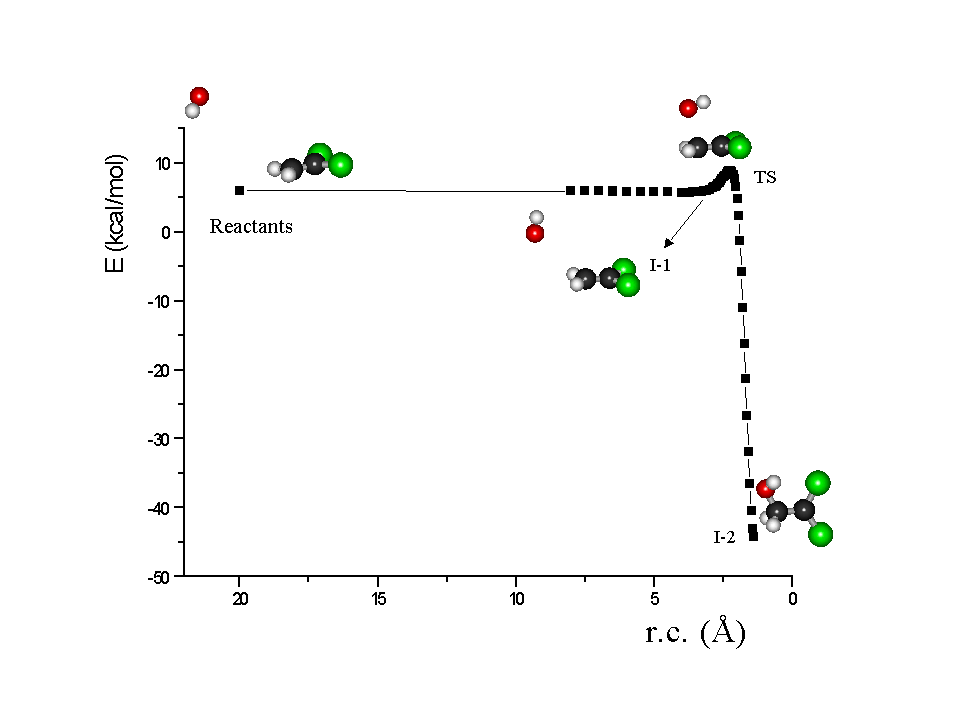
Figure 4: Reaction Coordinate for the reaction of OH + CH2=CCl2
as an example, calculated by PM3 method, where I-1 is a long-range complex,
T.S. is the corresponding Transition State and I-2 is the stable covalent
adduct or Intermediate.
From the computational analysis of the reaction pathways, we have estimated
the activation energies (Ea) and the total energies (DHr) of the proposed
reactions (Figure 4). It was not our aim, however, to determine accurate
activation energies for each reaction.
The reaction pathways were studied using semiempirical, PM3, and ab-initio
calculations (at UHF and B3LYP level of theory, with 6-31G** basis set).
With these semiempirical calculations it is possible to observe, with Br,
Cl and OH but not with O and NO3, the presence of a long-range complex
(I-1) prior to it been collisionally quenched to form a stable covalent
adduct (I-2). A similar trend for the Ea and DHr was observed between the
semiempirical and ab-initio calculations
Table 1: Activation Energies (Ea) (kcal mol-1) and Heat Contents
(DHr) (kcal mol-1) calculated by
PM3 method for the addition reaction of different electrophiles to chloroethenes.
|
C2H3Cl |
C2H2Cl2 |
Z-C2H2Cl2 |
E-C2H2Cl2 |
C2HCl2 |
C2Cl2 |
|||||||
| Ea | deltaHr | Ea | deltaHr | Ea | deltaHr | Ea | deltaHr | Ea | deltaHr | Ea | deltaHr | |
| O(3P) | 3.68 | -62.07 | 3.59 | -64.48 | 4.49 | -66.39 | 4.56 | -65.96 | 4.31 | -52.07 | 5.29 | -56.61 |
| OH | 3.76 | -47.84 | 3.07 | -50.20 | 4.74 | -49.69 | 4.71 | -49.25 | 4.21 | -50.63 | 4.31 | -50.52 |
| Cl | 0.10 | -37.15 | 0.03 | -39.05 | 0.50 | -34.05 | 0.94 | -34.44 | 0.81 | -36.74 | 1.10 | -35.12 |
| Br | 0.05 | -25.25 | 0.37 | -27.70 | 0.49 | -23.29 | 0.18 | -23.23 | 0.47 | -24.52 | 1.45 | -23.82 |
| NO3 | 3.16 | -43.98 | 3.37 | -43.19 | 2.86 | -39.29 | 2.73 | -38.60 | 0.22 | -40.83 | 1.53 | -38.41 |
In Table 1 the activation energies for the addition reactions of the different
electrophiles to the chlorinated ethenes are shown. The Ea, in general,
increase with the increasing chlorine substitution, although for 1,1 dichloroethene,
which is the most reactive chloroethene, the addition reaction has the
lowest Ea value.
Calculated heats of reaction DHr are also presented. All the addition reactions
studied are exothermic, with DHr generally decreasing with increasing chlorine
substitution.
Conclusions
Semiempirical quantum mechanics appears as a reasonable starting point
to establish reactivity trends of similar types of reactions and it is
possible to demonstrate that in this series of chloroethenes there is a
strong correlation of the rate constant with the atomic orbital coefficients
of the reactants that can be well explained by frontier molecular orbital
theory. Certainly to obtain more accurate values of the geometries and
the energies it is necessary to consider higher levels of ab-initio calculations.
For the chloroethenes studied in this work, the HOMO is composed carbon-carbon
p bonding and halogen atom lone-pair contribution. As a result, the rate
constants for these reactions do not correlate in a simple manner with
the energy of the HOMO (the experimental ionization potential), as in the
case in the other alkene reactions. A better description of this effect
could be achieved, taking into account the perturbation term which carries
the contribution of the different atomic orbitals to the HOMO of the chloroethene
through the atomic orbital coefficients.
Atmospheric implications
The rates of the tropospheric reactions of the alkenes and, in particular,
haloalkenes with OH, NO3, Cl, etc. are important compared with those of
other saturated volatile organic compounds. For the haloalkenes, reactivity
decreases with increasing substitution by halogen atoms in the molecule.
Of all the chloroethenes, only C2Cl4 has an atmospheric lifetime which
allows its presence in the upper troposphere where photolysis might take
place, thus contributing to stratospheric ozone depletion through catalytic
cycles.
In general, rapid removal of haloalkenes in the troposphere through reaction
with OH and other radicals could lead to a decrease in tropospheric radical
concentrations, thus having an indirect impact on stratospheric ozone through
enhanced catalytic destruction.
References
Y. Yokouchi, H. Akimoto, L.A. Barrie, J.W. Bottenheim, K. Anlauf and B.T. Jobson; J. Geophys. Res., 99, 25379, 1994.
J.H. Seinfeld and S.N. Pandis; Atmospheric Chemistry and Physics; John
Wiley and
Sons, N.Y. 1998.
M.A. Teruel, R.A. Taccone and S.I. Lane; Int. J. Chem. Kinet., 31, 867, 1999.
F. Kaufman; Progress in Reaction Kinetics; 1, 3, 1961.
M.D. King, C.E. Canosa-Mas and R.P. Wayne; Phys. Chem. Chem. Phys. 1, 2231, 1999.
I. Fleming; Frontier Orbitals and Organic Chemical Reactions, John Wiley
and Sons,
Chichester, 1996.
Back to
| Session 1 : Stratospheric Processes and their Role in Climate | Session 2 : Stratospheric Indicators of Climate Change |
| Session 3 : Modelling and Diagnosis of Stratospheric Effects on Climate | Session 4 : UV Observations and Modelling |
| AuthorData | |
| Home Page | |