 |
Stratospheric Processes And their Role in Climate
|
||||||||
| Home | Initiatives | Organisation | Publications | Meetings | Acronyms and Abbreviations | Useful Links |
![]()
|
Stratospheric Processes And their Role in Climate
|
||||||||
| Home | Initiatives | Organisation | Publications | Meetings | Acronyms and Abbreviations | Useful Links |
![]()
David W. Fahey, NOAA Aeronomy Laboratory, Boulder, USA (dfahey@al.noaa.gov)
Klaus Pfeilsticker, University of Heidelberg, Germany (pf@uphys1.uphys.uni-heidelberg.de)
A. R. Ravishankara, NOAA Aeronomy Laboratory, Boulder, USA (ravi@al.noaa.gov)
Reactive nitrogen oxides play key roles in the photochemistry of the upper troposphere and lower stratosphere (UT/LS). Significant progress has occurred in the last decades in both the measurement and modelling of reactive species and their distribution in the UT/LS. The sampling of the UT/LS has increased with the addition of new instruments on space borne and airborne platforms. Models have advanced to three-dimensional chemical-transport models and more precise and accurate trajectory models. Significant advances have also been made in quantifying rate coefficients of gas-phase, heterogeneous, and photochemical processes in the laboratory. This workshop was convened to address the recent progress in measurements, and modelling and to highlight areas of research in which significant uncertainties remain.
The Workshop was convened by G. Amanatidis (European Commission, Brussels, M. Chipperfield (University of Leeds, UK), R. A. Cox (University of Cambridge, UK), D. W. Fahey (NOAA Aeronomy Lab, Boulder, USA), N. Harris (EORCU, University of Cambridge, UK), Y. Kondo (University of Tokyo, Japan), M. J. Kurylo (NIST/NASA, Washington, USA), M. Pirre (CNRS Orléans, France), A. R. Ravishankara (NOAA Aeronomy Lab, Boulder, USA), through the initiative of Klaus Pfeilsticker of the University of Heidelberg who was also the local host. Approximately 65 people attended the three-day meeting on the University of Heidelberg campus. The meeting was organised with a core group of presentations followed by extended discussion sessions. Separate breakout sessions were held for modelling, observations, and laboratory measurements, and the meeting concluded with a rapporteur's report and summary for each of these topics.
In the following sections, the discussions and presentations of modelling and observations of reactive nitrogen in the UT/LS are summarised. The discussions and presentations identified areas of high confidence, areas of limited confidence, and knowledge gaps/needs as shown in Table 1.
Table 1. Knowledge summary**
|
Lower Stratospheric Modelling
|
Upper Tropospheric Modelling
|
Stratospheric and Tropospheric Observations
|
| - High Confidence | ||
|
|
|
| - Limited Confidence | ||
|
|
|
| - Knowledge Gaps/Needs | ||
|
|
|
| **Areas noted under Limited Confidence and Knowledge Gaps/Needs for Stratospheric and Tropospheric Modelling represent observational needs which are not repeated in the Observation entries. | ||
The principal component species of NOy in the stratosphere are HNO3, NOx, N2O5, ClONO2, BrONO2, and HO2NO2. NOx is here defined as the sum of NO and NO2. Figure 1 shows the photochemical interrelationship of these species. The most significant source of stratospheric NOy is N2O in reaction with O(1D) to produce NO and photolysis of NO is the primary in situ loss process of NOy. Contributions from galactic cosmic rays, solar proton events and solar electrons are relatively small (<20%) on average. In the lowermost stratosphere, lightning and aircraft NOx may contribute to the NOy budget. NOy is produced in the mid- and upper stratosphere and transported poleward and downward into the lower stratosphere (LS). NOy is chemically conserved in the LS. The main sink of NOy in the LS is cross-tropopause transport. Sedimentation of polar stratospheric cloud particles in the polar vortex can effect redistribution of NOy within the LS and transport NOy to the troposphere.
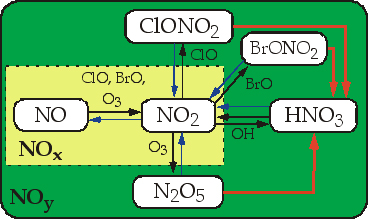 |
|
Figure 1. Schematic of reactive processes in the stratospheric NOy reservoir:
gas-phase (black), photolytic (blue), and heterogeneous (red). From Fahey and Ravishankara (1999).
|
|
(Please contact the SPARC Office, for a better resolution of the image)
|
Modelling NOy in the LS involves processes on very different time scales. The distribution of N2O and NOy depends largely on global transport, while, for example, the partitioning of NOx species has a much shorter time scale and can in many cases be considered to be in photochemical steady state. The mean age of air in the high-latitude stratosphere is several years or more. The partitioning between NOx and HNO3 takes place on an intermediate time scale of about a few days. Modelling the distribution of NOy thus requires 2D or 3D photochemical-transport models, while the partitioning within the NOy family can often be calculated by photochemical box models. Box models constrained by observations provide probably the best way to test our understanding of the NOy chemistry [see e.g. Gao et al., 1999; Cohen et al., 2000]. In many cases, however, not all relevant parameters needed to constrain the model calculations are measured sufficiently. In these cases, global 3D photochemical-transport models constrained by meteorological analysis provide the best tool available to interpret these measurements.
Many aspects of the stratospheric NOy chemistry are well understood. In particular, the principal species which constitute NOy in the stratosphere are known (Figure 1). In situ measurements from the ER-2 aircraft in comparison with the Mk IV remote measurements have shown that the sum of the individual NOy compounds is in good agreement with total NOy [e.g. Sen et al., 1998]. It is unlikely that there are any significant unidentified NOy components in the stratosphere. However, many models show relatively large discrepancies for the distribution of NOy compared with observations, even when N2O is constrained. In addition there are relatively large discrepancies for NOy and N2O between different models. These discrepancies are likely due to the transport features of the model and possibly the NO loss processes rather than the production of NOy from N2O decomposition.
The partitioning on short time scales that controls the NO/NO2 ratio, for example, seems to be well reproduced by constrained models [e.g. Del Negro et al., 1999]. However, the agreement with observations may be less good for globally unconstrained models, as the NO/NO2 ratio depends critically on ozone and the NO2 photolysis rate. One issue here is the treatment of the albedo in global models. Sen et al. [1998] show good agreement between the observed N2O5 diurnal cycle and constrained model calculations. Similarly, Stimpfle et al. [1999] report reasonable agreement between observed and modelled ClONO2.
The partitioning between NOx and HNO3 in the LS depends critically on the aerosol surface area. The partitioning between NOx and HNO3 is fairly well simulated in models which are constrained by observed aerosol surface area. Recent changes in reaction rate recommendations [e.g. Brown et al., 1999a,b] have improved the agreement between observed and modelled NOx/NOy. Discrepancies are on the order of 10-20% at ER-2 altitudes (15 – 20 km) [Gao et al., 1999]. However, comparisons between observed NO2 from SAOZ balloon-borne measurements and three-dimensional models show that at other altitudes the agreement is less good, with differences of the order of 50%. Jucks et al. [1999] have shown using far infra-red observations that constrained models underestimate NOx and overestimate HNO3, with the discrepancy increasing with altitude. One way to resolve this discrepancy would be to increase the HNO3 photolysis near 200 nm. It remains an open issue how well models reproduce the UV flux at short wavelength in the stratosphere. Many models include parameterisations of the atmospheric transmission in the Schumann-Runge bands, which have not been optimised for HNO3 photolysis calculation.
A critical parameter for modelling the partitioning between NOx and HNO3 is the concentration of OH. Both loss and production of HNO3 depend on OH. Previous studies have shown good agreement between observed and modelled OH, in particular for the ratio of OH/HO2. However, the most recent JPL 2000 reaction rate recommendations lead to large differences between modelled and observed OH/HO2 ratio, with observed OH about 50% higher than modelled [Lanzendorf et al., 2001]. Current models significantly overestimate the ratio of HO2NO2 to NOx for high-latitude spring, compared to observations. This may represent a gap in our current knowledge. A speculative long-wavelength photolytic pathway for HO2NO2 has been suggested to resolve this discrepancy [Wennberg et al., 1999].
Bromine nitrate (BrONO2) is an important bromine reservoir and thus a critical component for modelling stratospheric bromine chemistry. The heterogeneous hydrolysis of BrONO2 affects the partitioning of stratospheric NOx and HOx [e.g., Erle et al., 1998]. Unfortunately, BrONO2 has never been measured in the stratosphere. The observed rapid increase of HOx after sunrise is basically consistent with model calculations of the hydrolysis of BrONO2 and subsequent HOBr photolysis and may thus provide some indirect evidence for BrONO2.
PAN is generally not included in stratospheric models. Its contribution to NOy is believed to be small in the stratosphere but more significant in the tropopause region.
There was relatively little discussion at the workshop on formation of polar stratospheric clouds (PSC) and denitrification. Although models are able to predict some aspects of PSCs reasonably well and there has even been some success in modelling observed denitrification, many aspects of PSC formation are still uncertain. Very little is known about the precise PSC nucleation mechanism. This limits our confidence in modelling denitrification and the ability to predict the future evolution of Arctic ozone depletion in the presence of long-term trends of temperature and water.
In the UT, NOx and NOy essentially are governed by many processes that are highly coupled (see Figure 2). As in the stratosphere, the concept of NOy is a useful constraint and diagnostic. However, UT NOy is more variable due to the variability and uncertainty in emissions, chemical processing, transport, and loss by wet and dry deposition. Therefore in the UT, it is often more important to measure and model individual species such as NOx, HNO3, and PAN. The principal tropospheric NOy species and their interconversion processes are shown in Figure 3.
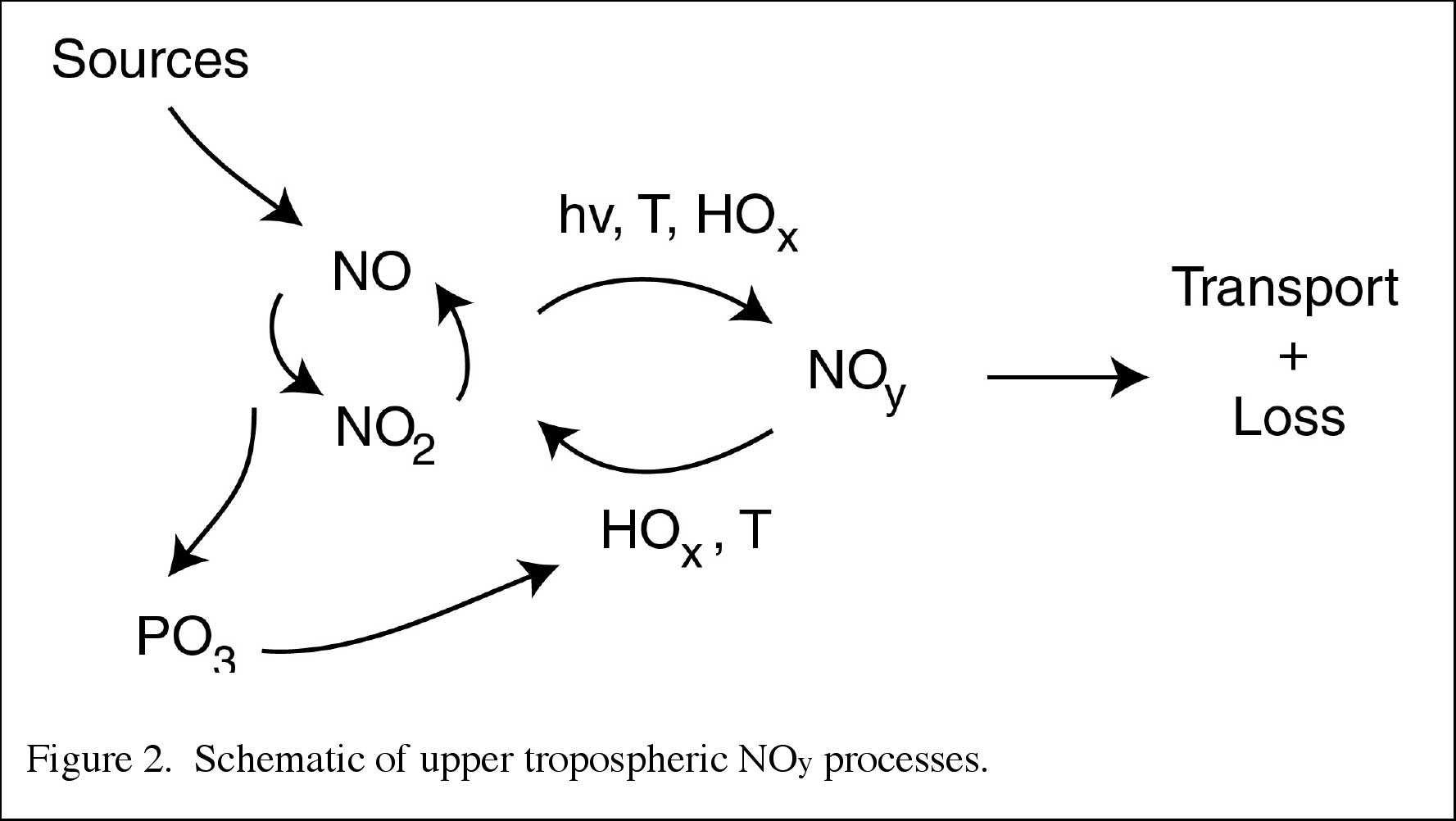 |
|
Figure 2. Courtesy of K. Law
|
|
(Please contact the SPARC Office, for a better resolution of the image)
|
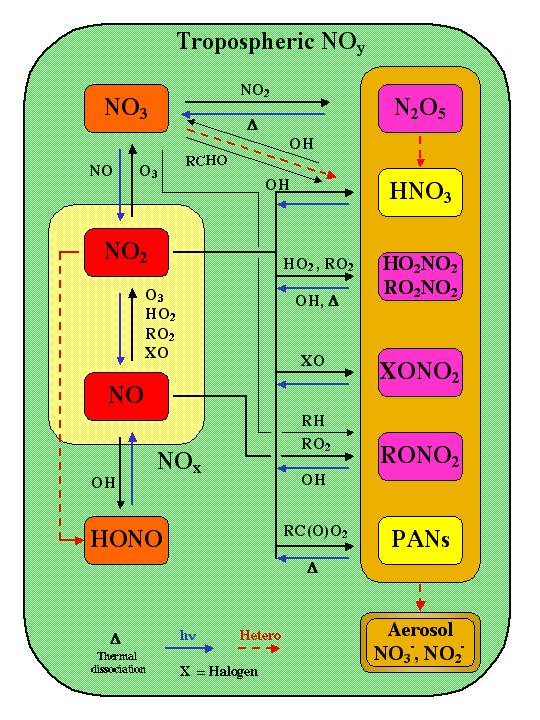 |
|
Figure 3. Schematic of tropospheric NOy species and their interconversion processes.
Courtesy of B. Ridley
|
|
(Please contact the SPARC Office, for a better resolution of the image)
|
By contrast to the stratosphere, where chemical and transport time scales are often very different, both transport and photochemistry act on the distribution of NOx, HNO3, PAN, and other NOy compounds in the UT with similar time constants of weeks to months. Mixing ratios of UT NOx and HNO3 can be lower than those at the surface and in the LS. Cross-tropopause transport and convection are sources of NOx and HNO3 in the UT. Direct aircraft emissions of NOx into the UT are also important, as is NOx generated by lightning in the UT or at lower altitudes where it can be transported to the UT by convection. Net removal of NOy compounds from the UT is primarily by conversion to HNO3 followed by uptake into particles and sedimentation. One consequence of the coequal time scales for transport and photochemistry is that comparatively few species are routinely in 24-hour photostationary state within the UT. Diagnostics, such as the number of condensation nuclei or the abundance of other short-lived species, have been suggested as indicators of the residence time of air in the UT.
In the UT there are still many uncertainties and unknowns related to our understanding about NOx and NOy and associated chemistry. Two areas were identified where our knowledge is felt to be good. These include our understanding of fast gas-phase photochemistry involving HOx and NOx partitioning where the use of observationally constrained box models run to steady state has shown that we have a reasonably good understanding about some aspects of radical budgets in the UT. However, uncertainties still remain about the exact nature of HOx sources and sinks and the rate of in-situ O3 production at high NOx levels [see for example, Jaegle et al., 1999]. There is a lack in our ability to simulate NOy partitioning, most notably the NOx/HNO3 ratio. Although several mechanisms have been proposed (e.g. reactions on soot) to address the discrepancy between observed and modelled ratios (the latter are too low), no satisfactory explanation has been found to date. It was also felt that we have reasonably high confidence in the total amount of NOx emitted by aircraft.
With respect to NOx emissions, the distribution of lightning emissions in mid-latitudes is reasonably well known [Huntreiser et al., 1998; Dye et al., 2000]. However, large uncertainties remain in describing the distribution of lightning in the tropics and the magnitude of NOx emissions from this source, particularly in the tropics. Also, there are considerable difficulties in modelling this source due to the scales involved and the coupling to convection. The problem of including emissions, which are very heterogeneous in time and space, in large-scale global models which have grid boxes on the scales of 10-100s km is a problem which requires further attention, in general, particularly as the response of the chemical system is non-linear with respect to NOx concentrations. Global inventories do exist for surface and aircraft emissions and the total amounts of NOx emitted from some sources (aircraft, combustion) are well known compared to those from other sources (biomass burning, soils). However, several data sets currently used by models require updating to more accurately represent emissions in 2000, for example. It was also felt that the stratospheric flux of NOy into the troposphere is reasonably well known.
Modelling NOx and NOy in the UT (and elsewhere) relies on accurate kinetic and photolysis rate calculations. There is a clear need for more accurate data on reaction rates, cross sections and quantum yields under UT temperature and pressure conditions and for the mechanisms, processes, and rates of heterogeneous reactions. Related to the latter is the whole question of how to simulate heterogeneous processes in models. This topic is one that will require much more attention in the future. Also, whilst models are able to reproduce measured photolysis rates in clear sky conditions, there are still considerable problems reproducing data collected in cloudy and high aerosol conditions. This will require both new data and model improvements.
The ultimate fate of NOy in the free troposphere involves washout of soluble species, such as HNO3, uptake on aerosols/droplets/ice particles or transport downward to the surface and loss by dry deposition. These processes can also affect the partitioning between NOx and NOy, and are especially important if they regenerate active NOx which is then available to produce O3 in the troposphere. The whole question of loss of HNO3 and other species by washout has received too little attention both in the modelling community and the measurement community largely due the complexity of the processes involved. Indeed, the treatment of clouds in models is still very crude and the treatment of wet deposition depends mainly on derived precipitation rates together with information on solubility. Measurements of nitrate in rainwater have been used to validate modelled wet deposition of HNO3 but clearly more observational data and also laboratory data on rates and mechanisms are still needed.
The relative distributions of NOx and NOy in the UT depend on transport processes as well as emissions, chemical processes, and loss processes. Based on the comparison of global models with (rather limited) tracer data (e.g. CFC-11) and the improved reliability of current weather forecasting models, it can be concluded that our ability to model the large-scale transport in the troposphere is reasonably good. This also explains why back trajectory calculations have been used very successfully in recent years to study the origin of different air masses observed during measurement campaigns. However, many transport processes occur at smaller scales. These include frontal transport and stratosphere-troposphere exchange which are resolved by current models when run at high enough resolution although convection embedded in frontal systems and sharp gradients around the tropopause are often not resolved. Deep convection is parameterised in current chemistry-transport models. Whilst there have been improvements in recent years (e.g. see results from WCRP model intercomparison exercises, e.g. Jacob et al., 1997), further improvements can be envisaged based on new parameterisations, increased resolution, and the use of nested models. These will clearly be necessary if we are going to be able to simulate fine-scale layers that are widely observed in the UT. At the present time it is unclear whether there is a need to model these features in order to simulate the global NOx, NOy, and O3 budgets more accurately. Further measurements of tracers that are very useful for validating transport processes in models would significantly improve our modelling capability (e.g. 222-Rn, 210-Pb, CO2, 7Be/10Be).
Finally, as a way forward, the idea of a formal model/data intercomparison was discussed. State of the art box models, including current chemical schemes could be compared to available NOx and HOx data from various airborne campaigns. Comparison with NOy data and its individual components would require the use of global or regional models that take into account transport and scavenging processes. A main objective would be to test the sensitivity of current models to these processes over a wide range of conditions in the UT.
There is high confidence that the distribution of NOy within the LS is controlled almost entirely by transport from the middle and upper stratosphere and across the tropopause. The NOy created at high altitudes within the stratosphere is then distributed into the LS by advection and diffusion on a time scale of years. In the overworld (defined as the stratosphere above the 380K potential temperature surface), there are extensive, high-resolution in situ observations of N2O, the principal source of NOy. Based on these observations, we have a good working knowledge of the N2O distribution in space and time. In the overworld, observations have demonstrated that there is a tight correlation of NOy with N2O, except in air that has been influenced by recent PSC sedimentation, and that this correlation varies seasonally. For N2O values less than about 50 ppbv, the NOy/N2O correlation becomes non-linear and is associated with greater uncertainty.
By contrast to the distribution of NOy which is set on a time scale of years, the partitioning of NOy among the different species, HNO3, NO, NO2, N2O5, ClONO2, etc., within the LS is established on a time scale of minutes to 30 days. Because transport of air in the stratosphere is approximately zonal, most of the photochemical relationships between NOy species exist in a 24-hour diurnal photostationary state. The most important NOy species and all the gas phase processes that interconvert them have been identified (see Figure 1).
Some observations have been analysed to show that we have sampled a broad range of one or two of the parameters controlling the NO/NO2 ratio and the NOx/HNO3 ratio, but a narrow range in others. Where we have sampled a broad range in the parameters, we can provide a quantitative evaluation of the completeness and accuracy of model processes. For example, atmospheric observations at low temperatures imply that the uncertainty of the rate coefficient for NO+O3 is 5-15% at 200K and by less than that over the range 200-220K. This uncertainty is significantly less than that currently derived from the extrapolation of laboratory measurements made at higher temperatures. Observations also indicate the rate ratio of the OH + NO2 reaction to the sequence of processes that convert NO2 to N2O5 and then to HNO3 by heterogeneous reaction are accurately represented in models using current laboratory rate expressions.
While we have high confidence in our knowledge of the distribution of NO, NO2, NOy, observations of NO3, N2O5, HNO4, PAN, ClONO2, and HNO3 are far fewer, and for the most part these observations have been made by remote sensing and lack the context of simultaneous, accurate free-radical measurements. Consequently the confidence in our knowledge of the distribution of these species and the chemistry affecting that distribution is limited. This is primarily important for HNO3 because it can condense at very low temperatures. For the other species, the lack of observational tests means that our ability to demonstrate that we have identified all of the important processes and established that their rates are correctly modelled is not currently possible.
Observations of the composition of the middle-world (below 380K) are also not as extensive as observations of composition in the overworld. Our ability to describe the frequency and intensity of cross-tropopause transport and subsequent mixing on the composition in this region is limited.
An increasing trend has been observed in column NO2 amounts over the last two decades. The most likely cause is related to NOy aerosol chemistry or stratospheric transport. Concerning NOy, we have little to no understanding or observational demonstration of whether or not trends in tropospheric N2O are producing the expected NOy trends in the stratosphere. Combined long-term records of NO2, HNO3, aerosol, N2O, and the extent of denitrification are needed. Observations should include sampling in the tropics, mid-latitudes, and the poles and at altitudes capable of resolving exchange between the UT and LS across the tropopause.
Although the new SOLVE observations [e.g. Fahey et al.] demonstrated the existence of a class of very large PSC particles with strong denitrification potential, information of their nucleation are still lacking. Additional studies confirming the generality of the new observations should be conducted.
There is considerable room for improvement in our understanding of the effects of stratospheric transport on the distribution of NOy. Much of this is not specific to NOy but associated with clear problems that have been identified in our ability to model the mean age and the age spectrum of lower stratospheric air. The mean age has been derived from the correlation of N2O with quite extensive measurements of CO2 and SF6, and the age spectrum from the CO2 measurements. For NOy specifically, observations and analyses of observations that would lead to an improved understanding of the seasonal cycle of NOy and its relationship to both the mean age and the age spectrum in middle-world is needed.
The role of NOy production by high-energy electrons associated with geomagnetic disturbances is unclear. Since the geomagnetic disturbances are closely related to the 11-year solar cycle, this production mechanism may cause a similar cycle in NOy in the LS.
Comparison of HNO3 chemistry in the lower and upper stratosphere points to possible errors in descriptions of the photon flux at short UV wavelengths (< 310 nm). These could potentially be resolved by in situ actinometry to measure JO2, JNO, JN2O or JHNO3.
The use of chemical co-ordinates should be expanded to more precisely refine our understanding of fast photochemistry and to more clearly define which aspects of stratospheric NOy science are dependent on transport and which on chemical parameters alone.
Aerosol surface area is a central control over the abundance of NOx in the LS. Demonstration of a model capable of predicting from elementary principles the observed aerosol-N2O correlations and/or the geophysical distribution of aerosol remains a challenge.
Balloon-borne, long-path UV-visible measurements have occasionally shown relatively high levels of NO2 together with high levels of OClO in the Arctic vortex. An analysis shows that these observations are not consistent with our current understanding of winter stratospheric chemistry. Further work will examine the role of vertical resolution and spatial inhomogeneity on the retrievals.
The chemistry of the UT is more complex than the chemistry of the stratosphere because many more species are involved and the system is inherently much more heterogeneous with orders of magnitude variation routinely observed in NOx, hydrogen radical precursors and H2O. The sampling of the UT has increased significantly in the last decade due to an increased number of airborne sampling missions using instrumented aircraft in the US, Europe, and Japan; and due to the successful launch of new satellite instruments.
Airborne platforms are now frequently instrumented for a wide suite of reactive nitrogen measurements including NOy, NO, NO2, HNO3, and PAN and occasionally for other organic nitrates. The sampling campaigns of these aircraft have made measurements over a wide range latitudes, longitudes, and seasons. Based on a number of airborne field programs, it is felt that the principal components of NOy are generally well described in the UT largely because discrepancies between total measured NOy and the individual components are often rather small. The minor species however are not known with high precision. Extensive measurements of NOx have been made in concert with HOx and other species to constrain the rate of ozone production in the UT. Based on a model constrained by extensive UT measurements, the loss rates of NOx are found generally to exceed the production rates in large remote regions of the UT. Other measurements have been used successfully to quantify NOx sources in the UT, including lightning and aircraft emissions. The observations of NO2 obtained from a new space borne instrument and of NOx made on board commercial aircraft demonstrate the enormous potential of global scale measurements to lead to improved understanding of reactive nitrogen processes. A number of published studies [e.g. Emmons et al., 1997] have compiled NOx and NOy data from one or more airborne campaigns and made these first-order climatologies available to the atmospheric sciences communities. A range of studies has used these data sets to demonstrate the skill of multidimensional models of the UT.
In order to improve our understanding using models, more extensive measurements in the UT are still needed. Specifically, more comprehensive measurements suitable to constrain NOy production and loss processes would help define missing chemistry or unexpected uncertainties in accepted kinetic or photolytic rates. New measurements should also include minor NOy species such as HO2NO2 and organic nitrates.
The current state of our understanding of various reactions important in controlling the partitioning between NOx and NOy, and determining the abundance of the individual nitrogen oxide species were discussed. In addition, some key atmospheric reactions of species such as OH, HO2, and ClO that affect NOx and NOy abundance were discussed in the workshop; however, they are not highlighted below. Most of the key reactions coupling reactive nitrogen species within the NOy reservoir have been measured in the laboratory and evaluated by the NASA Data Panel [NASA, 2000] and the IUPAC Panel [IUPAC]. The reactions discussed below and summarised in Table 2 are those considered to be of particular importance for further study based on their role in NOy photochemistry and the uncertainty associated with the reaction.
Table 2. Summary of priorities for future laboratory studies
| Gas-phase processes |
|
| Photochemical Processes |
|
| Heterogeneous Processes |
|
To validate atmospheric chemistry models in the UT/LS, it was suggested that rate constants should be determined to better than 15% accuracy. The general feeling was that this goal is presently difficult to achieve for many of the reactions involving NOy species.
The most important gas-phase radical reactions for further study were determined based on their importance in the atmosphere or the large uncertainty associated with their rate constants. Those of high priority include the reactions of OH with NO2 and HNO3, the reaction of O with NO2, the reaction of NO with O3, and the reactions that lead to the formation and destruction of RO2NO2 molecules. A major photochemical process in the atmosphere associated with large uncertainty is the overtone photolysis of HO2NO2. These processes are very briefly discussed below.
• OH + NO2 -> Products (1)
Calculating ozone production is highly sensitive to the value of the rate coefficient for this reaction, which is one of the most investigated reactions. There is a wealth of data at 300K spanning more than four-orders-of-magnitude in pressure [NASA, 2000; IUPAC]. Currently, it is suspected that this reaction consists of two competing channels that result in HNO3 and peroxynitrous acid (HOONO) [Golden and Smith, 2000]. To date, HOONO has not been detected. The work of Hippler and co-workers on this reaction has provided evidence for the existence of an unstable intermediate thought to be HOONO [Hippler et al., 2001]. The O-O bond dissociation energy in HOONO is estimated to be 20.1 kcal mol-1. Hippler et al. have fitted the available data at 300 and 400K as a function of pressure to this two-channel mechanism. The limiting high-pressure rate constants are consistent with the isotope scrambling experiments of Donahue et al. [2001] but are somewhat different from the interpretations of Golden and Smith [2000]. The interpretation of Hippler et al. implies that one has to be careful in ascribing the measured OH temporal profile to either the sum of the two channels or to just one channel, because the assignment critically depends on the lifetime of HOONO relative to the OH observation time. The detection of HOONO, measurement of the rate coefficients for its formation, and interpretation of the existing data in terms of the two possible channels awaits further work.
• The temperature dependence of O + NO2 and NO + O3
The reaction O + NO2, the rate limiting step in the NOx-catalysed ozone destruction, was identified as an area of emerging confidence. Recent work, along with the some of the previous measurements, have better defined this rate coefficient and shown that it is ~15-20% higher at lower stratospheric temperatures than currently accepted values. The negative temperature dependence, albeit small, suggests that the reaction is not a simple abstraction reaction.
For the rate coefficient of the NO + O3 reaction above 308K, the Arrhenius plot curves upwards such that a linear extrapolation of the high temperature data to lower temperatures becomes uncertain. This is further complicated by the possible differing rate coefficients for the reactions of the two spin-orbit states of NO with ozone and the changes in their relative abundance with temperature. Additional high quality data down to 200K, where the current uncertainty is approximately a factor of two [IUPAC, NASA, 2000], is desirable.
• The equilibrium of peroxynitrate species at atmospherically relevant temperatures
With the view that peroxynitrates are atmospheric reservoirs of NO2, the equilibrium constants for reactions of the type RO2NO2 <- -> RO2 + NO2 are needed over a large temperature range to accurately predict the atmospheric fate of RO2NO2. RO2 radicals may be both alklyperoxy and acylperoxy radicals. Both forward and reverse reactions involving CH3O2, C2H5O2, and CH3C(O)O2 and the thermal decompositions of CH3C(O)O2NO2 and C2H5C(O)O2NO2 have been studied. Future studies should extend the temperature range for forward and reverse reactions, obtain absolute rate constants for these reactions, and obtain kinetic data as a function of pressure to separate the low- and high-pressure rate constants.
. OH + HNO3
This reaction has recently been extensively studied by Brown et al. [1999a,b; 2001] and has been assessed by the NASA panel. However,
an independent confirmation of the Brown et al. results is warranted. The rate coefficient, which increases with decreasing temperature, is pressure
dependent. The only products are H2O and NO3, even though reaction proceeds via the formation of a complex,
HNO3•OH, that is long enough lived to make the rate coefficient pressure and bath-gas dependent at low temperatures.
The following reactions also were found to have high priority for future studies:
NO3 + NO2 <- -> N2O5
HO2 + NO2 <- ->HNO4 (PNA, peroxynitric acid, see above)
RO2 + NO <- -> ROONO (peroxynitrite).
Other important atmospheric reactions that indirectly impact the NOx to NOy ratio and the abundance of the NOx and NOy species, and require
further study were found to be:
CH3O2 + HO2 -> CH3OOH + O2
OH + CH3OOH -> products
Formation and fate of CH3C(O)OOH (acetylhydroperoxide).
OH + O3, HO2 + O3, HO2 + NO, HO2 + OH reactions.
• J(HNO4) in the infrared region
Overtone photolysis of HO2NO2 was highlighted in this workshop. Even though the photodissociation cross section in the near infrared spectral range may be quite small, the rate of this process may be significant owing to the large actinic flux in this spectral region. The need for further laboratory work on the photochemistry of HO2NO2 to reduce the uncertainty in the UV and IR absorption cross sections was stressed. A new value for the standard enthalpy of formation of HO2NO2 of –52.7 ± 8 kJ mol-1, compared to the old value of –57.24 kJ mol-1, shifts the dissociation threshold from 8787.2 to 8407.4 cm-1. However, this change is not sufficient to explain the photochemical action spectrum obtained at –20°C that showed photodissociation activity around 7000 cm-1 [Wennberg et al., 1999]. However, this results needs confirmation.
Other photochemical processes that need further study were identified to be the photochemistry of PAN and photolysis quantum yield of CH2O.
• The heterogeneous interaction of HNO3 with water ice
The gas phase lifetime of HNO3 strongly depends upon the coefficient, g, for the uptake on cirrus clouds. The value of g is constant at 0.3 in the range 170 to 195K and decreases with temperature between 195K and 240K [IUPAC, NASA, 2000]. Certain uptake experiments of HNO3 on H2O-ice substrates simulating cirrus clouds have resulted in unlimited uptake up to a temperature of 210K whereas other experiments resulted in significant saturation of the HNO3 uptake below 210K [Zondlo et al., 2000]. Currently, the temperature above which saturation of the ice surface by HNO3 occurs is uncertain.
Several workers have proposed a significant decrease of the H2O evaporation rate from ice upon HNO3 uptake [Livingston and George, 1998; Warshawsky et al., 1999] and thereby extending the lifetime of cirrus clouds. In contrast, such an effect was not observed in another study [Beirmann et al., 1998]. A decrease of the H2O evaporation rate due to the presence of adsorbed HBr [Hudson et al., 2001] and HCl on ice [Delval et al., 2001] has also been obtained. No definite assessment of the role of adsorbed HNO3, HCl, and HBr on ice may be given at this time with regards to the resulting lifetime of atmospheric ice particles. Quantification of the changes in H2O evaporation rates due to HNO3 uptake as a function of temperature is needed. In addition, the nature of the coating with respect to the evaporation rate of H2O on ice needs to be carefully assessed in view of several thermodynamically stable or metastable structures.
• Reactions that may convert HNO3 back to NOx
A few reactions were considered that might convert HNO3 back to NOx and, thereby, alter the NOx to NOy ratio in the UT. They include the reaction of CH2O with HNO3 to give HCOOH + HONO in sulphuric acid aerosols and the possible reduction of HNO3 to NO2 in droplets. Even though the first reaction has been studied a few times, more quantitative information is needed to assess its role in the atmosphere.
• Surrogate surfaces for heterogeneous reactions
In general, laboratory studies of heterogeneous reactions have been carried out using surrogates for atmospheric substrates. It has recently become clear that structural and chemical features of the condensed phase can control both the kinetics of the uptake and the identity of the products. Furthermore, the gas uptake itself may alter the surface and the bulk substrate. Substrates of interest include ice, soot, and mineral dust. The soot generated in the laboratory may not represent all of the features of atmospheric soot. Laboratory studies alone are not sufficient to derive the properties of environmental soot particles; field measurements are badly needed. In the absence of field characterisation, soot with differing properties have been generated by adjusting the combustion conditions from fuel lean to fuel rich.
Another drawback of using the surrogates is the correction for the porosity of the substrate. In a number of cases the correction for porosity has been included without experimental verification of whether or not such an application of pore diffusion theory was justified. For “sticky” molecules, whose desorption rate constant is significantly slower than the rate of surface reaction, it is expected that pore diffusion will not be important because the reaction takes place before the molecule has a chance to explore the internal surface. Conversely, in the case of a fast desorption rate, one needs to correct the observed rate of uptake because of an enhanced gas-surface collision rate.
Many other issues were briefly discussed but they are not included in detail here. The discussed topics include coating of combustion aerosol (soot) particles by sulphuric acid; producing internal and external mixtures of aerosols and their surrogates for laboratory studies [Jacobson, 2001]); and dealing with mineral dust particles in the context of laboratory studies, and identifying the best model substrates for laboratory studies.
Biermann, U. M., J. N. Crowley, T. Huthwelker, G. K. Moortgat and P. J. Crutzen, Geophys. Res. Lett. 25, 3939-42 (1998).
Brown, S. S., J. B. Burkholder, R. K. Talukdar and A. R. Ravishankara, J. Phys. Chem. A 105, 1605 (2001).
Brown, S. S., R. K. Talukdar and A. R. Ravishankara, J. Phys. Chem. A 103, 3031-3037 (1999a).
Brown, S. S., R. K. Talukdar and A. R. Ravishankara, Chem. Phys. Lett. 299, 277-284 (1999b).
Cohen, R. C., et al., J. Geophys, Res. 105, 24283-24304 (2000).
Del Negro, L. A., et al., J. Geophys, Res. 104, 26687-26703 (1999).
Delval, C., B. Flückiger and M. J. Rossi, in preparation (2001).
Donahue, N. M., R. Mohrschladt, T. J. Dransfield, J. G. Anderson and M. K. Dubey, J. Phys. Chem. A 105, 1515-1520 (2001).
Dye, J. et al., J. Geophys, Res. 105, 10023-10045 (2000).
Emmons, L. K. et al. Atmos. Environ. 31, 1851-1905 (1997)
Erle, F., U. Platt, K. Pfeilsticker, Geophys. Res. Lett. 25, 4329 (1998).
Fahey, D.W. et al. Science 291 1026-1031 (2001)
Fahey, D. W. and A. R. Ravishankara, Science 285 208-210 (1999)
Gao, R. S., et al., Geophys. Res. Lett. 26, 1153-1156 (1999).
Golden, D. M. and G. P. Smith, J. Phys. Chem. A 104, 3991-3997 (2000).
Hippler, H., St. Nasterlack, F. Striebel and D. M., Golden, Workshop on nitrogen oxides in the lower stratosphere and upper troposphere, University of Heidelberg, Heidelberg, Germany, 19-22 March 2001, p. 47.
Hudson, P. K., K. L. Foster, M. A. Tolbert, S. M. George, S. R. Carlo and V. H. Grassian, J. Phys. Chem. A 105, 694-702 (2001).
Huntrieser, H., H. Schlager, Ch. Feigl, H. Höller, J. Geophys. Res. D 103, 28247-28264 (1998).
IUPAC, International Union of Pure and Applied Chemistry, Subcommittee for Gas Kinetic Data Evaluation. http://www.iupac-kinetic.ch.cam.ac.uk
Jacob, D. J., et al., J. Geophys, Res. 102, 5953-5970 (1997).
Jacobson, M. A., Nature 409, 695-697 (2001).
Jucks, K. W., et al., J. Geophys, Res. 104, 26715-26723 (1999).
Lanzendorf, E. J., et al., Geophys. Res. Lett. 28, 967-970 (2001).
Livingston, F. E., and S. M. George, J. Phys. Chem. A 102, 10280-88 (1998).
Jaeglé, L., et al., Geophys. Res. Lett. 26, 3081 (1999).
NASA, Panel for Data Evaluation, Chemical kinetics and photochemical data for use in stratospheric modelling, JPL Publication 00-003, 2000. http://jpldataeval.jpl.nasa.gov
Sen, B. et al., J. Geophys, Res. 103, 3571-3585 (1998).
Stimpfle, R. M., et al., J. Geophys, Res. 104, 26705-26714 (1999).
Warshawsky, M. S., M. A. Zondlo and M. A. Tolbert, Geophys. Res. Lett. 26, 823-26 (1999).
Wennberg, P. O., et al., Geophys. Res. Lett. 26, 1373-1376 (1999).
Zondlo, M. A., P. K. Hudson, A. J. Prenni and M. A. Tolbert, Ann. Rev. Phys. Chem. 51, 473-99 (2000) and references therein.
Rapporteurs: R. Cohen, R. S. Gao, K. Law, M. Rossi and B.-M Sinnhuber
Back to SPARC Newsletter 18 Homepage
![]()